The prion agent
There has been much speculation as to the molecular nature of the infectious agents responsible for these disease conditions. In 1972 Stanley Prusiner proposed that these agents were not viruses but a unique class of infectious agent totally free of nucleic acid and consisting solely of protein. Prusiner coined the term prion (from proteinaceous infectious particle) to describe the agent and was awarded the Nobel Prize in 1997 for his work investigating prions and TSEs. Evidence for the proteinaceous, nucleic acid-free nature of the agent has come from a number of experiments in which the chemical and physical nature of the infectious agents has been examined. They are resistant to heat inactivation at temperatures that would readily destroy DNA or RNA and are resistant to both ultraviolet light and ionizing radiation, treatments that normally damage microbial genomes. No susceptibility to DNAse, RNAse or enzymatic hydrolysis is detectable but the agents are sensitive to treatment with urea, SDS, phenol, and other protein-denatur- ing chemicals. These characteristics indicate that the prion has, as proposed, a protein- aceous character lacking an infectious or transmissible nucleic acid.
Of course all proteins are encoded by nucleic acids and the prion protein (PrP) is no exception, being encoded by a cellular gene, the Prnp gene. Found in all mammalian and bird genomes examined to date the protein is a glycoprotein of 208 amino acids that cycles between the endosomal and plasma membranes of various cell types, including neuronal cells. The biological function of the cellular protein, PrPc, is unknown although evidence suggests a role in transmembrane transport or signaling. Mice lacking this protein develop normally, suggesting redundancy in its function and critically such mice are also resistant to infection by disease-forming prion proteins (designated PrPsc taken from the disease scrapie), indicating the requirement for the cellular protein in disease. How do PrPc and PrPsc differ and how is the disease state achieved? The answer lies in the different conformations of the two proteins; the PrPc has a high proportion of alpha-helical structure but can undergo a conformational change to the pathogenic PrPsc form, which has reduced helical structure as regions refold into beta-sheets. This altered form of the protein shows increased resistance to proteases and reduced plasma membrane expression within the cell and, more importantly, increased self-aggregation, forming long fibrils of
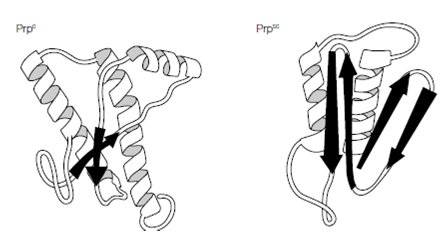
Figure: Conformational changes in PrP.
protein that eventually become insoluble, causing the cellular damage. How PrPc converts to PrPsc and whether the process is reversible is unclear, but there seems strong evidence that the presence of PrPsc ultimately converts more and more PrPc until the cell is destroyed. The concept of prion protein conformational conversion supports the three disease routes observed with TSEs. Inheritance of a mutated Prnp gene can lead to familial (hereditary) disease, a spontaneous transcription error during gene expression would give sporadic disease and acquisition of the PrPsc isoform initiates transmissible disease.
As with conventional microbes there are different strains of prion that appear to ‘breed true’ in terms of the characteristic symptoms they produce following infection of new hosts. For example, scrapie can be transmitted to both sheep and goats where, despite variations in the amino acid sequence of their PrP proteins, it displays identical disease characteristics. Likewise some strains of BSE can be propagated by several animal species (each with their own different prion protein) and the three-dimensional conformation of the refolded PrPsc is identical regardless of the host species. These findings suggest that the final structure of PrPsc and its disease characteristics are independent of the host. By contrast, transmission of scrapie (via contaminated feed) into cattle gives distinct characteristics, and confirms that the contribution of infecting and host prion proteins is not yet understood.
In humans there are two common versions (polymorphisms) of the prion protein that differ in a single amino acid (valine or methionine at coding position 129). There are also rare mutant forms, many of which are associated with inherited susceptibility to prion disease. The polymorphic variation at position 129 does not itself lead to disease, but influences the clinical characteristics of a disease-promoting mutation that can occur at position 178. Mutation of the normal aspartate to asparagine at this position leads to CJD if there is valine at position 129, but to fatal familial insomnia (FFI) if the individual pos- sesses methionine at amino acid 129. Sheep likewise have several different forms of the prion protein linked to susceptibility, whereas cattle have two forms that do not appear to be associated with susceptibility to BSE.